StarDrop is an application from Optibrium that was designed to aid decision making for scientists involved in drug discovery.
- Virtual Library Enumeration – The Nova plug-in module for StarDrop now has the added ability to quickly and easily enumerate a virtual library based on a template scaffold that you define with substitution points and variable fragments. You can sketch the groups to substitute at each point, select them from a user-defined or centrally administered library, or take them from a decomposition of another series using the R-group analysis tool in StarDrop
- Data visualisation – now allows you to apply interactive filters to your graphs and plots to quickly focus on the most interesting compounds. StarDrop now also supports the analysis of dates allowing you to explore variations of properties or scores with time
- Clustering – this new tool enables you to easily identify groups of similar compounds within a data set, based on either their structural similarity or properties
- Dataset Filtering – this helps you to remove compounds from a data set with unwanted sub-structures or property values. You can define any number of criteria with which to filter a data set
- Duplicate Removal – when combining compound data from multiple sources it’s common to end up with multiple copies of the same compound in a single data set. The duplicate removal tool makes it easy to find these and choose the entries that you want to keep.
- ADME QSAR – new model for predicting log([Brain]:[Blood]) (the old model remains available for consistency with previously calculated results)
- StarDrop now includes a FieldAlign module, using Cresset’s molecular Field technology, provides a unique, 3-dimensional (3D) insight into the biological activity, properties and interactions of your compounds.
Virtual Library Enumeration
The Nova module allows you to generate and evaluate new chemistry ideas either by enumerating a virtual library or now by applying a chemical transformation to a chemical template. The resulting molecules can then be scored against a target activity and calculated physicochemical properties or off-target activities.
A detailed explanation can be found in this publication, Applying Medicinal Chemistry Transformations and Multiparameter Optimization to Guide the Search for High-Quality Leads and Candidates”, Segall et al., J. Chem. Inf. Model., 2011, 51 (11), pp 2967–2976, DOI,
There are over 200 chemical transformations already defined and you can pick and choose which you want to apply to the template molecule.
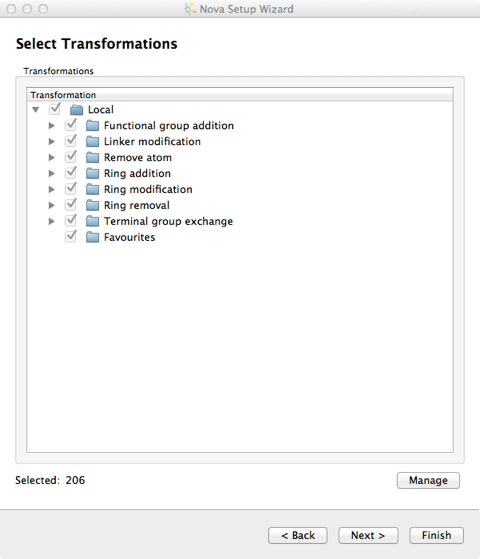
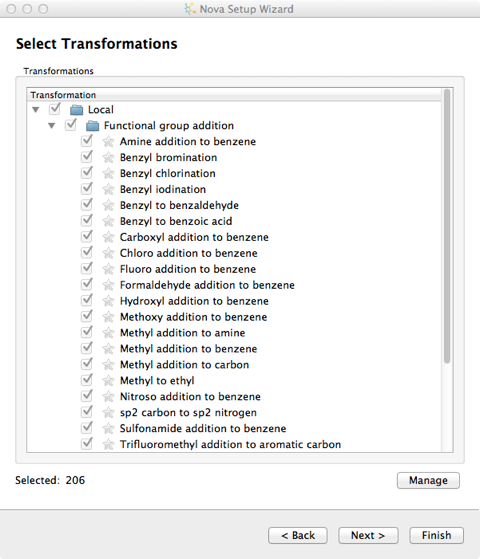
New transformations can be imported into the user’s local library from the “Transformations” button on the Nova tab and additional libraries can be specified in the Nova tab of the Preferences dialogue, from the File->Preferences menu option. Transformations are imported in SMIRKS format, SMIRKS is a reaction transform language.
The file format is a tab delimited text file and the name of the file is used to name the new group.
|
1 2 |
SMIRKS Description Reference |
Example:
|
1 2 3 |
[C:2]1[C:3][CH:4]=[N:5][C:6]1.[N+:7]#[C-:8].[C:10](=[O:11])[O:12][H:88]>>[H:88][N:7][C:8](=[O:12])[CH:4]([C:3][C:2]1)[N:5]([C:10]=[O:11])[C:6]1 Ugi, I; Meyr, R.; Fetzer, U.; Steinbrückner, C. (1959). "Versuche mit Isonitrilen". Angew. Chem. 71 (11): 386. [C:1]=[C:2][C:3]=[C:4].[C:5]=[C:6]>>[C:1]1[C:2]=[C:3][C:4][C:5][C:6]1 DielsAlder Diels, O. .; Alder, K. . (1928). "Synthesen in der hydroaromatischen Reihe". Justus Liebig's Annalen der Chemie 460: 98–122 |
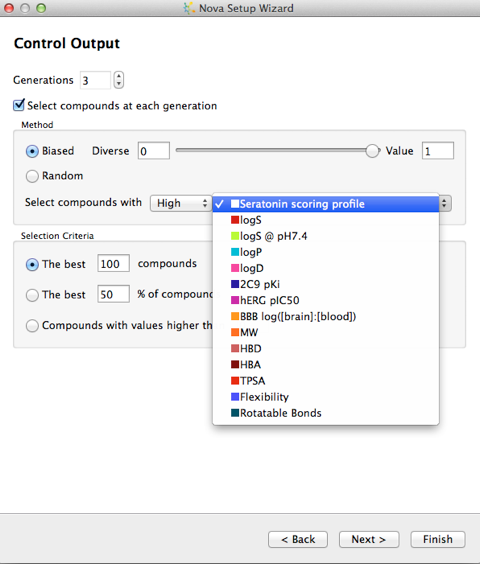
The next step allows you to control the building process, if you choose multiple generations then there is a real risk of a combinatorial explosion in the number of molecules generated, the best way to control this is to select a subset of the generation to be used in the next iteration of transformations. There are several options to control the output, either a random selection or based on a calculated property.
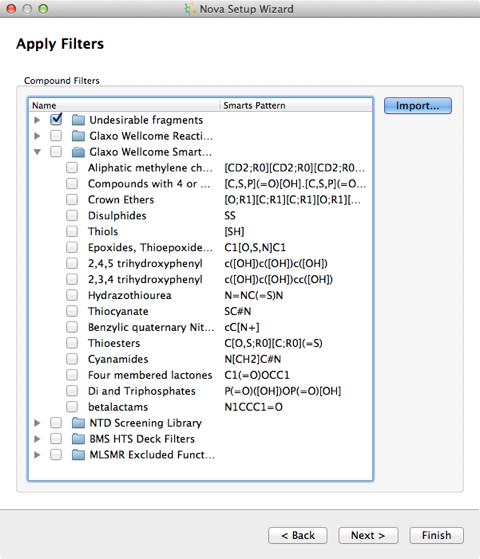
The final step is the option to filter the output, you can indicate any structural features which you would like to ensure are not present in any of the new compounds. Compounds that contain any of these will be removed at the end of the process. A number of published sets of filters are available and by clicking the Import… button you can add your own. To define your own you must create a text file containing SMARTS and their associated names. The SMARTS patterns must not contain any spaces and there should be a space to separate the pattern from the name, with one pattern and name on each line of the file.
Example:
|
1 2 |
[S,C](=[O,S])[F,Br,Cl,I] acid halide |
On my MacBook Pro (2.6 GHz Intel Core 2 Duo, 4 GB 667 MHz DDR2 SDRAM) and taking a single molecule as the starting point and applying 50 transformations for 3 generations, filtering by predicted bioactivity resulted in the generation of 240 molecules in less than a minute.
By selecting one of the resulting structures you can see the parent structure from which it was derived by transformation, and any children generated in subsequent generaations.
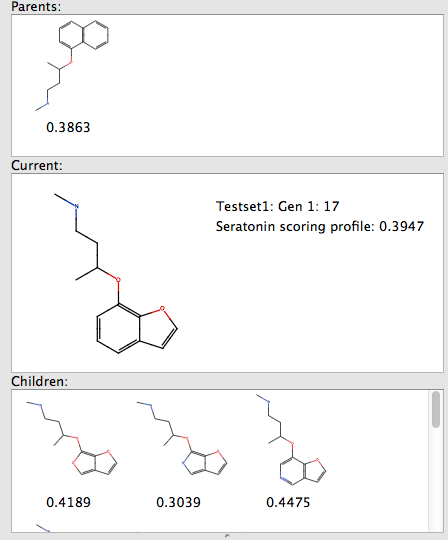
The speed of calculation means that users will very quickly become accustomed to trying out ideas without worrying about bogging the computer with seemingly never ending calculations. Once you are happy with the generated molecules you can of course then submit them to FieldAlign.
ADME QSAR
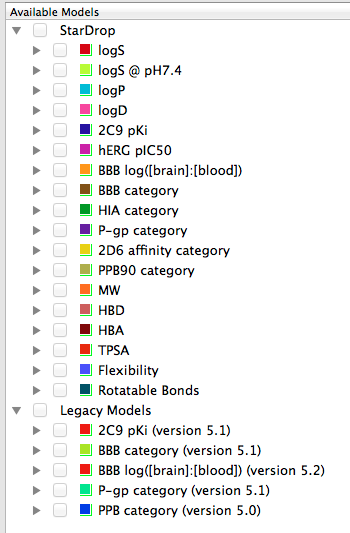
StarDrop provides a number of physicochemical property calculations including Molecular weight, Hydrogen bond donors, Hydrogen bond acceptors, Topological polar surface area, Flexibility and Rotatable bonds.
In addition there a number of more sophisticated calculations available.
logP (Octanol/Water) logD7.4 (Octanol/buffer at pH 7.4) Solubility – Aqueous Solubility – Solubility in PBS at pH 7.4 Human Intestinal Absorption CNS (Blood-Brain Barrier) Penetration Cytochrome P450 Affinities – CYP2C9 – CYP2D6 P-gp Transport hERG pIC50 Plasma Protein Binding
Some of the models have been updated but if you want to compare with previously calculated molecules the old models are still available under “Legacy Models”
FieldAlign Integration
FieldAlign is a software package developed by Cresset that given the 3D structure of your active compound will use it to align a series of compounds (2D or 3D) with unknown activity based on calculated field points, optionally a protein binding site or excluded volumes can be included. Firstly the generation of multiple conformers is automatically handled by FieldAlign, using a combination of torsion driving and high-temperature dynamics, followed by a short minimisation step to the nearest local minimum. Field points are then generated using Cresset’s method which relies on XED models of atoms and molecules, which present a more complex, accurate description of the charge around any atom. Then each of the conformations is compared with the known active and a similarity score generated. The results are passed to StarDrop and extra properties can be calculated. There is a review of FieldAlign here.
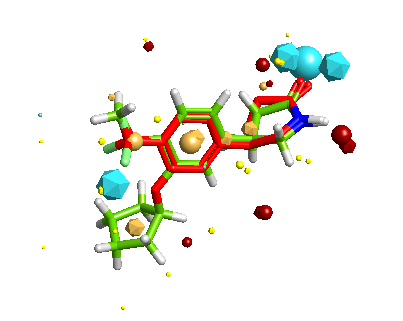
The inclusion of FieldAlign adds a very powerful new tool and allows the scoring of the 3D structures and the involvement of target protein information in the scoring of molecules.
There is a minor graphics glitch with FieldAlign on a small number of Macs with a specific graphics card, I am assured this will be corrected in a mid-year update.
Summary
StarDrop is a very interesting application, the desktop provides useful tools for analysing data and generating a selection of important descriptors and provides a competent selection of visualisation tools but structure-based searching is limited. Linking to the server however provides an extra powerful dimension to the tool providing access to sophisticated computational models. The ability for novice users to generate models of their data will be very much appreciated by those who have been daunted by R, expert users will also appreciate being able to easily disseminate their results for use by other users. The extension of the Nova module allows the building of novel molecules in a chemically intelligent manner rather than a blind combinatorial enumeration. The addition of FieldAlign adds a very important new dimension to the application.
Last Updated 9 April 2013