The molecular operating environment MOE is an integrated molecular design and modelling platform that handles Small Molecules – Peptides – Biologics.
I’m very grateful for Chemical Computing Group for providing a M1 version of MOE and license key to test, and for the support providing helpful comments and suggestions.
Since I wanted to test the multiprocessor support I started MOE using on the M1 machine
$MOE/bin/moe -mpu 10
First read in 3g6d.pdb.gz this is virtually instantaneous on both systems.
Then run QuickPrep to prepare the structure
The Intel MacBook Pro took 1 minute 23 seconds
The M1 MacBook Pro took 41 seconds.
Then annotated (Antibody| IMGT), selected 3 residues as shown below and then ran a residue scan
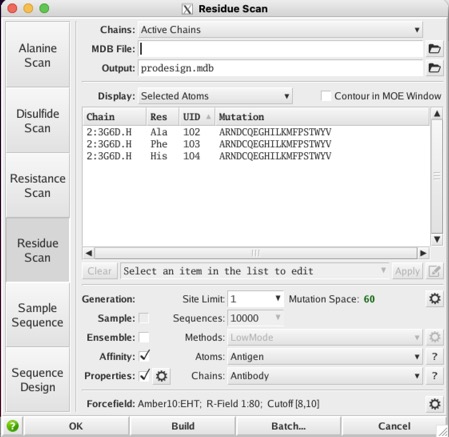
I took the average of 3 runs
The Intel MacBook Pro took 16 minutes 28 seconds
The M1 MacBook Pro took 6 minute 47 seconds.
Identical molecules SVL script
dbnbmols_incommon.svl calculates a matrix for the number of compounds each database has in common with each of the other databases based on either structure or fingerprints.
I used 26 databases of commercially available fragments that were loaded into individual MOE databases, the SVL script was then loaded and the command below used.
SVL command used
|
1 2 |
db_nb_mols_incommon [dbv_KeyList[], 'MatrixOut.txt', [write_cpds:1, cpds_outfile:'ident_cpds.txt']] |
The Intel MacBook Pro took 6 minutes 35 seconds
The M1 MacBook Pro took 4 minute 14 seconds.
Importing files into MOE database
I imported a selection of structures from ChEMBL, 2D structures in sdf file format. MWt 250 to 500, calc LogP 0 to 5. This is a 2.6 GB file containing 1,144,624 molecules.
The Intel MacBook Pro took 13 mins
The M1 MacBook Pro took 5 mins
Processing sd Fileș
MOE contains a collection of applications specially designed for processing structure data (SD) files from the shell command line.
sdwash applies washing rules and tautomer enumeration to structures in SD files. For testing I used a selection of structures from ChEMBL, 2D structures in sdf file format. MWt 250 to 500, calc LogP 0 to 5. This is a 2.6 GB file containing 1,144,624 molecules
Command used
|
1 2 |
time sdwash -pH 7 -salts ChEMBLsubset.sdf -o output.sdf |
The Intel MacBook Pro took 6 hours 11 mins
The M1 MacBook Pro took 3 hours 24 mins
Using moebatch
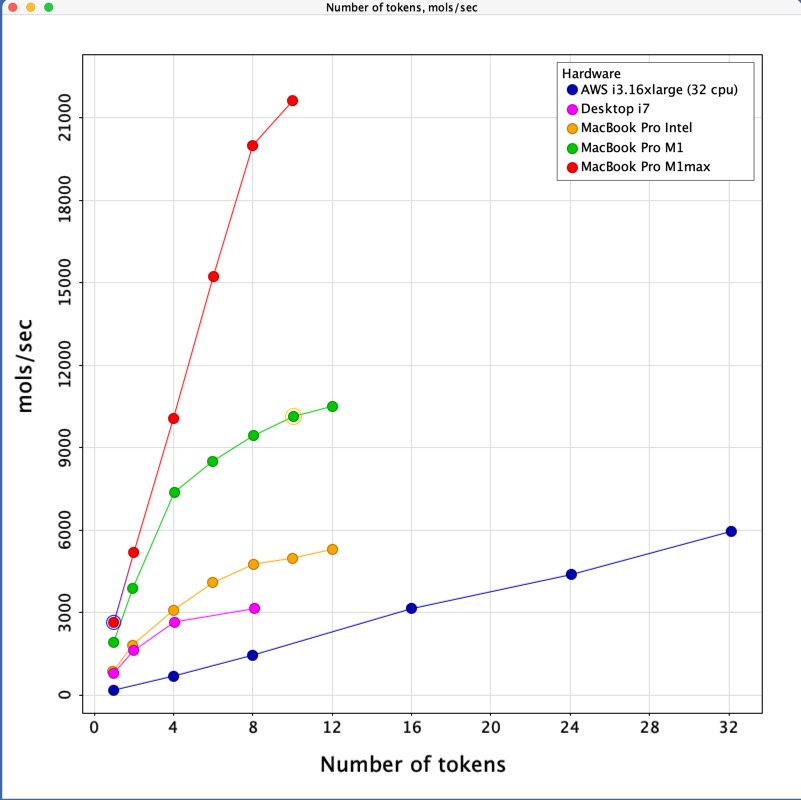
Pharmacophore searching is a critical part of virtual screening and is can be used to search very large datasets. Pharmacophore query generation in general requires user interaction and is not well-suited for batch mode. Pharmacophore search, on the other hand, can be done in MOE/batch using an SVL script or runnable file that invokes the SVL function ph4_Search. The search was run on a dataset of 274K structures using a predefined query. CCG also provided me with timings from other architectures and the results are shown below. As you can see the MacBook Pro M1 max out-performs all other platforms tested, this is particularly noticeable in the multicore performance, evaluating over 20,000 molecules per second. Whilst the fans came on after a minute or two with the other machines they remained silent on the MacBook Pro M1 max.
Building combinatorial libraries
There is a lot of interest in virtual screening of large virtual libraries of molecules. There several options for building these large libraries, it is possible to try and enumerate all possible molecules that conform to valency rules and do not include chemically unstable functional groups, the drawback is that it generates molecules that can be challenging (or impossible) to make. An alternative is to use reaction-based methods to combine known building blocks and reagents using a database of known chemical reactions. This approach whilst limited to the database of reactions at least provides a starting point for synthesis.
There are two options for reaction-based library enumeration in MOE, under “Fragments” or “QuaSAR”. The one under Compute | Fragments does not support multi-processing while the one under Compute | QuaSAR does support multi-processing.
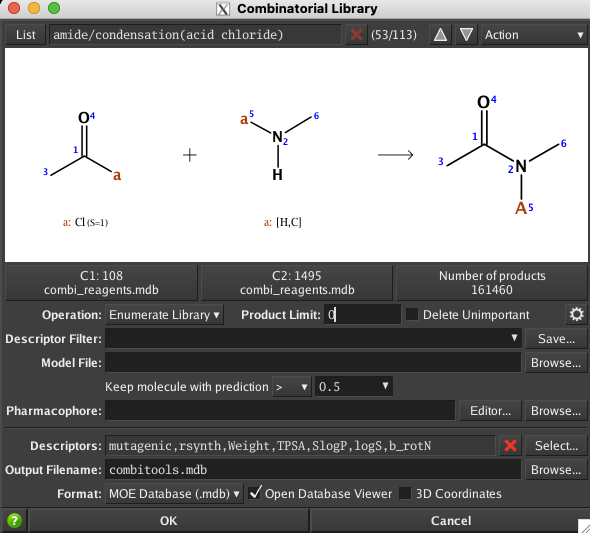
t is pretty straight-forward to take the output from one reaction as the input for the next step.
On the Apple MacBook Pro M1max I built a 4.5 million molecule library in 1.5 hours which is pretty impressive. Unfortunately the process did not finish on the Intel machine due to a power cut, however based on how far it had got I guess it was 5-10 times slower.
List of tools tested https://macinchem.co.uk/software-reviews/cheminformatics-and-compchem-on-apple-silicon/
Last update 25 Feb 2022