The 2018.01 release of Chemical Computing Group’s Molecular Operating Environment (MOE) software includes a number of new features, enhancements and changes.
Mac OSX 10.6 and Windows XP have been removed from the list of officially supported platforms. Supported Windows platforms are Vista/7/8/10, and the minimum supported MacOS is 10.7 (Lion).
There is also support for Hewlett-Packard Zvr Virtual Reality Display. This looks really neat but unfortunately I’ve not been able to try it out 🙁
Enhanced Display
One of features that I think I’ll be using most is the new 2D overlay, for example if you have a protein structure containing an active site ligand bound, it can sometimes be difficult to work out the structure of the ligand. The new 2D button in the MOE Footer opens the 2D overlay.
This gives a 2D depiction of ligands in the system overlaid on a corner of the MOE Window as shown below.
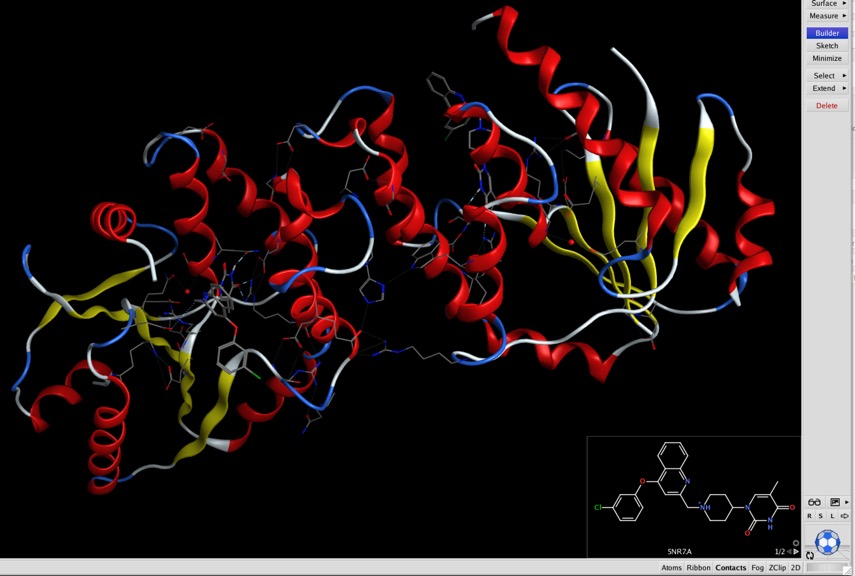
The display is interactive, such that any selection in either the 2D or the 3D display immediately highlights the corresponding atoms in the other display. One really useful aspect of this is that you can select ligand atoms in the 2D display, change colour, rendering etc. without worrying about accidentally selecting atoms in the protein.
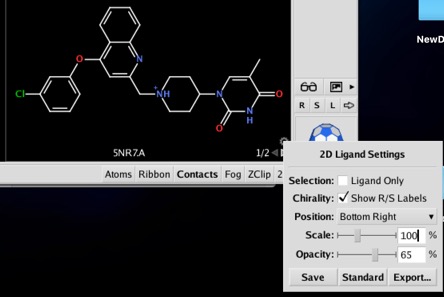
Opacity, size, location, and chirality labeling are configurable by clicking on the unfilled circle
Torsion scan
This is something I’ve been asking for. A new application for performing torsion scans on small molecules has been added to MOE. The application calculates energy as a function of dihedral angle and can be used to assess conformational flexibility, estimate rotational barriers and identify preferred dihedral angles. The application can be started with MOE | Compute | Conformations | Torsion Scan
Database Functions
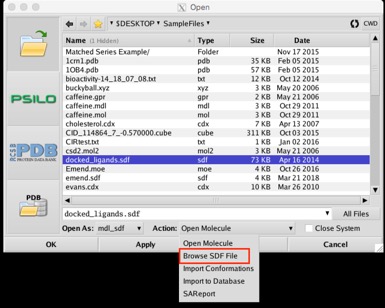
When opening SD files containing multiple molecules, an option is provided to browse the file contents rather than importing to an MDB or loading into MOE Keyboard up and down arrow keys can now be used to browse all lists in the Open panel, including the file list
The Database Viewer footer status line now features menus on each of its elements. Click on the words in the status line to open menus for entries and fields, including for just selected and visible. There is also a new spyglass icon opens the database search application.
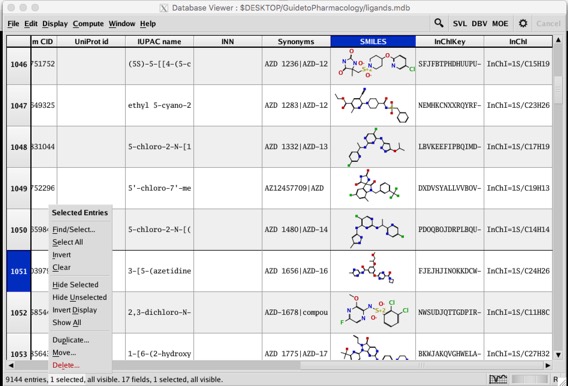
Annoyingly one bug has not been resolved, In the database viewer if the ligand ID is in the molecule field. when you minimise the structures the ligand ID is lost.
Building
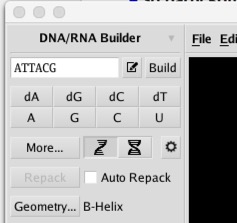
There is now a new DNA/RNA Builder. This new builder is similar to the Protein Builder in layout and functionality, and offers the following features:
- Build, append to, join, and split nucleic acid structures. DNA can be built as either a single helix or a double helix.
- Assign standard configurations. A, B, C, and Z helix geometries are supported.
- Mutate and explore nucleotide conformations. Convert between ribose and deoxy forms.
- Support non-natural base variants.
You can simply enter the DNA/RNA sequence as a text string and then click the build button
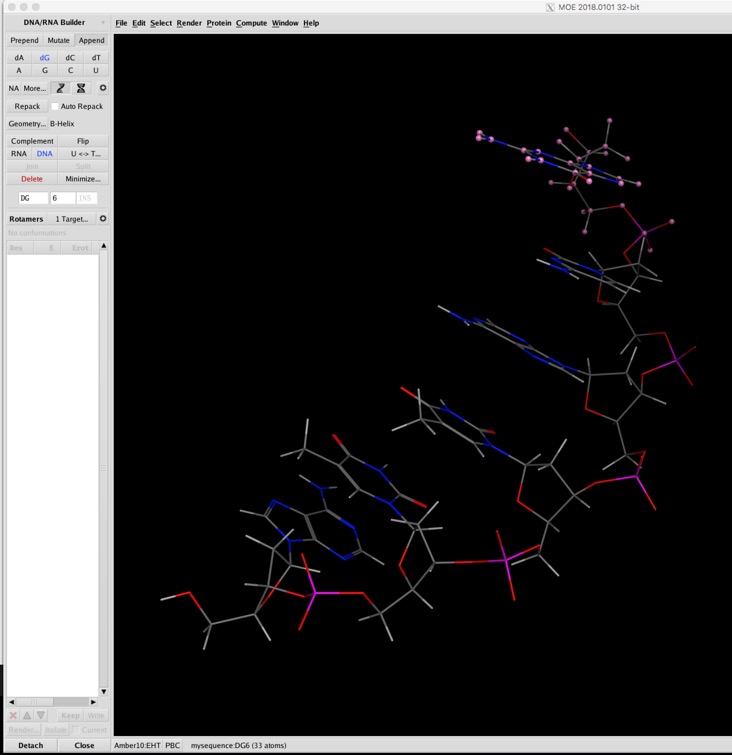
What is really nice is that with a click of a button the DNA can be built as either a single helix or a double helix. and similarly to protein alignment and superposition, similarity, RMSD, and other colour/measurement tools are available. In addition Protein-nucleic acid docking is supported under Protein-Protein docking in the Dock application.
I don’t use this but I notice the Antibody Modeler interface has been rewritten, featuring a simplified interface for automatic building.
Another feature I’ve been asking for is support for modern multi-core machines (without the need for additional licenses!) and I see several applications have been parallelized to run in the moe -mpu environment:
- Descriptor calculations with the SVL function QuaSAR_DescriptorMDB.
- Energy minimization in the Database Viewer DBV | Compute | Molecule | Energy Minimize.
- Conformational search using MDB input files in MOE | Compute | Conformations | Search.
- Rotamer library generation with DBV | Compute | Build Rotamer Library.
- Project database creation with the SVL run file dbupdate.svl and the scripts $MOE/bin/projupdate and $MOE/bin/projupdate.bat.
This is a great start, I can see why additional licenses might be needed to run on a cluster but not on my desktop machine.
The Amber14 parameter set is now supported in MOE. The new parameters consist of improvements to nucleic acids; otherwise, protein and small molecule parameters (and charges) are unchanged, in addition MOE now provides an interface to the AMBER program through the Dynamics panel MOE | Compute | Simulations | Dynamics. The molecular system is prepared in the same way as for MOE or NAMD dynamics.
MOEsaic, the web-based application for Structure Activity Relationship (SAR) and Matched Molecular Pair (MMP) analysis, has been enhanced, R-group contributions to molecular properties can now be calculated. Searching and plotting have also been improved.
There are also many improvements to the protein modelling side, the Antibody Modeler interface has been rewritten and a new application for mapping epitopes or other protein-protein interfaces (PPI) has been added. A new MOE Project database containing T-Cell Receptor (TCR) – Major Histocompatibility Complex (MHC) x-ray structures has been added. Since protein-protein interactions are an increasingly important area of research these improvements are very timely.
Let Update 5 April 2018