I’ve been using the latest update to MOE from Chemical Computing Group for a while now and i thought I’d post a few impressions.
The published new features include:-
Streamlined Interactive Modeling Interface
• Toggle ligands, proteins and surfaces on/off • Analyze and optimize multiple ligand:receptor complexes • Create surfaces, calculate properties and display substitution points
Integration of NAMD Engine in MOE
• Export parameters and scripts automatically • Import NAMD trajectories into MOE database • Run simulations on a cluster with restart capability
Structure-Based Medicinal Chemistry Transformations
• Transform molecules in 3D using reaction style rules • Refine structures in an active site and apply 3D filters • Integrated with scaffold replacement, fragment linking, growing and BREED
Enhanced Graphics
• Faster real-time GPU ray-tracing • 3D Stereo with anaglyph glasses • Clip molecular surfaces only
Non-bonded Interaction Visualization
• Display H-bonds, CH…X, proton-π and VdW interactions • Show strengths or energies and set thresholds • Control visualization for ligand, receptor and solvent combinations
Kinase Database and Explorer
• Search database of 3D aligned kinase structures • Add in-house structures with automated protocol • Browse kinases by core, pocket or canonical structural views
Perhaps the most striking update is the graphics, the 3D graphics capabilities in MOE have been substantially updated to make use of modern Graphics Processing Units, in particular faster real-time GPU ray-tracing, 3D Stereo with anaglyph glasses, Clip molecular surfaces only. I’ve used the Crystal Structure of a Phenhexyl/Oxazole/Carboxypyridine alpha-Ketoheterocycle Inhibitor Bound to a Humanized Variant of Fatty Acid Amide Hydrolase (3K7F) to look at a few of the new features. The ligand and the overall structure are shown below.
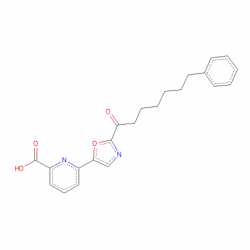
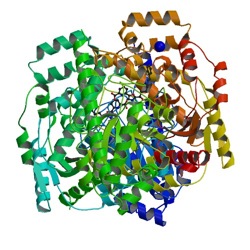
The binding of the ligand is interesting in that it nicely demonstrates a variety of different types of ligand-protein interactions. The 3D movie below was generated using ScreenFlow to capture the MOE screen. The movie (25MB) illustrates a number of the new features, the non-bonded interactions between the aromatic rings, the strength of hydrogen bonds shown by the size of the cylinder on the dotted line between interacting atoms, the ability to clip just surfaces and not the molecular structure and of course the 3D Stereo with anaglyph glasses. I tried a couple of different scheme and found the green/magenta gave the best 3D effect whilst supporting reasonable colours on the surfaces. You can get very inexpensive glasses here from Amazon 3D Glasses Green and Magentafrom 3DGB(http://www.3dgb.co.uk/), they also do clip on versions for users who wear glasses, or alternatively if you prefer Ebay
The MOE side bar has changed with a number of regularly used features that used to be buried in sub-menus now available directly, this should speed up interaction with the program (when I get used to it!), you should remember the menu layout can be modified using SVL and a number of “skins” are available to change the menu depending on the task in hand. The System button on the MOE Button Bar now raises a pop-up panel, shown in the movie (2MB) below, that provides instant control over the objects in MOE. All chains are grouped by chain tag into a collapsible listing. Within each tag group, the atoms are partitioned into Receptor, Solvent and Ligand classes and a control line presented. Each control line can affect the display of the atoms it represents: Select/deselect. Active/Inert without affecting visibility Hide/Show and simultaneously activate/deactivate Color Atoms (carbon atoms only or all atoms) Render Mode (point, line, stick, ball and stick, etc.) Hide/Show Surfaces associated with the atoms Delete atoms or surfaces or both
The MOE Footer now displays to the current selection (end of movie above) and provides information and controls for editing objects. Atom names, bond lengths, bond angles and dihedral angles can modified directly from the footer. In the case of dihedral angles, a wheel can be used to modify a torsion, and the Profile button displays a graph of energy vs. dihedral angle. Clicking in the plot will modify the structure. Unfortunately at least on my laptop some of the menus extend off the screen.
I’m not a big user of molecular dynamics but I suspect the integration with NAMD will be popular with a number of people. You can download the source code or precompiled binaries from the NAMD website(requires free registration). I put the binary in usr/local, remember to add it to your $PATH in your .bash_profile. I did a simple test and it seems to work as advertised. If you are new to molecular dynamics it might be worth installing VMD a free molecular visualiser and running through the tutorials.
This is a very useful update, the after using it for a few days I can see that the focus on improving work-flow actually does work and the 3D Stereo with anaglyph glasses certainly enhances presentations.