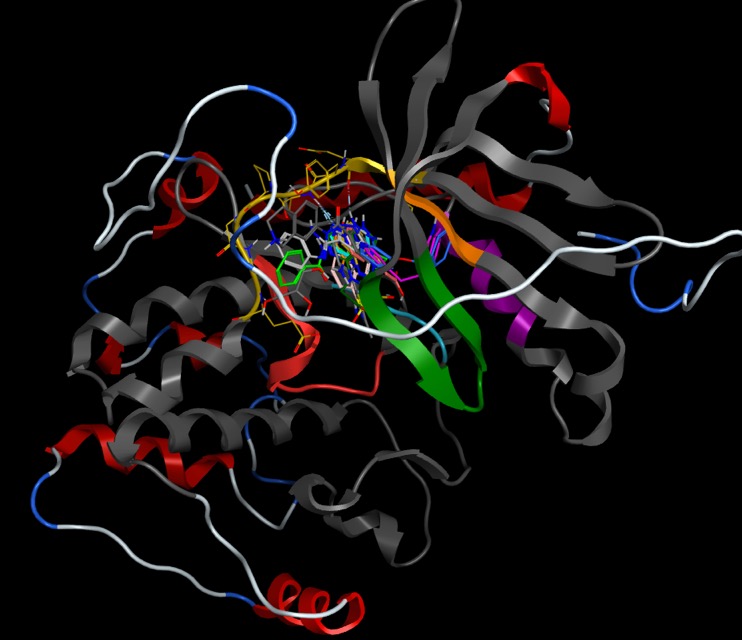
Much of our understanding of the binding of ligands to kinases has come from crystallographic studies, and there are currently several thousand kinase crystal structures in the PDB many for the same protein with different ligands bound. Protein kinases are enzymes involved in the transfer of the terminal phosphate of ATP to substrates that usually contain a serine, threonine or tyrosine residue. Kinases share a conserved arrangement of secondary structure elements that fold into a characteristic twin-lobed catalytic core structure with ATP binding in a deep cleft located between the lobes. The heteroaromatic adenine ring of ATP then forms a series of hydrogen bonds with the hinge region (shown in yellow below) between the two lobes of the protein.

Most of the inhibitors bind in the region of the ATP binding site (probably due to fact that most were discovered using enzyme assays) using the hydrogen bonding interactions of the hinge region. shown in the schematic below.

We can use the knowledge of these hing binding motifs to flag potential kinase inhibitors, as described in a publication by Xing et al. Scaffold mining of kinase hinge binders in crystal structure database DOI
Protein kinases are the second most prominent group of drug targets, after G-protein-coupled receptors. Despite their distinct inhibition mechanisms, the majority of kinase inhibitors engage the conserved hydrogen bond interactions with the backbone of hinge residues. We mined Pfizer internal crystal structure database (CSDb) comprising of several thousand of public as well as internal X-ray binary complexes to compile an inclusive list of hinge binding scaffolds. The minimum ring scaffolds with directly attached hetero-atoms and functional groups were extracted from the full compounds by applying a rule-based filtering procedure employing a comprehensive annotation of ATP-binding site of the human kinase complements. The results indicated large number of kinase inhibitors of diverse chemical structures are derived from a relatively small number of common scaffolds, which serve as the critical recognition elements for protein kinase interaction.
The structures of the most common hinge binding fragments are shown below.

Fragment-based screening has become an increasingly popular means to identify novel starting points for drug discovery programs, indeed 108 of the published fragment hits were identified when screening against kinases. Looking at the structures of the fragments identified it is clear they fall into a limited number of structural classes, the most common are shown in the scheme below.

All appear to bind to the ATP binding site, hydrogen bonding to the hinge region as shown below.

These fragments can all be included as SMARTS queries and used in a modified version of the match patters script. The results are shown below. Obviously the current limited list of SMARTS won’t include all possible hinge binding motifs and I’d be happy to modify them to include any contributions.

The Flag Potential Kinase Inhibitors Vortex Script
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 104 105 106 107 108 109 110 111 112 113 114 115 116 117 118 119 120 121 122 123 124 125 126 127 128 129 130 131 132 133 134 135 136 137 138 139 140 141 142 143 144 145 146 147 148 149 150 151 152 153 154 155 156 157 158 159 160 161 162 163 164 165 166 167 168 |
# Custom version of the Match Patterns script # This script flags molecules that contain a motif that may bind to the hinge binding region of kinases # Created by Macs in Chem https://www.macinchem.org import time import java from com.dotmatics.vortex.mol2img import Mol2Img from Queue import Queue from threading import Thread processorcount = java.lang.Runtime.getRuntime().availableProcessors() class smilesworker(Thread): def __init__(self, q, eval_column): self.q = q self.eval_column = eval_column Thread.__init__(self) def run(self): while 1: row = self.q.get() if row == None: return try: vortex_tmp_value = vortex.getMolProperty(vtable.getStructureText(row), "SMILES") except: vortex_tmp_value = None if (vortex_tmp_value == None): self.eval_column.setValueFromString(row, None) else: self.eval_column.setValueFromString(row, str(vortex_tmp_value)) #Kinase patterns here patterns = [ ["7-deazaadenine","Nc1ncnc2[nH]ccc12|HsExplicit"], ["Indazole","c1n[nH]c2ccccc12|HsExplicit"], ["2aminopyrimidine","[H]Nc1ncccn1|HsExplicit"], ["Adenine","Nc1ncnc2[nH]cnc12|HsExplicit"], ["2-Aminopyridine","[NH]c1ccccn1|HsExplicit"], ["[1,2,4]triazolo[4,3a]pyridine","c1nnc2ccccn12|HsExplicit"], ["3-aminopyrazole","Nc1cc[nH]n1|HsExplicit"], ["Imidazo[1,2a]pyridine","c1cn2ccccc2n1|HsExplicit"], ["4-Aminoprymidine","[NH]c1ccncn1|HsExplicit"], ["7-Azaindole","c1cc2cccnc2[nH]1|HsExplicit"], ["2-Aminopyrazine","[NH]c1cnccn1|HsExplicit"], ["3amino1Hpyrazin2one","NC1=NC=C[NH]C1=O|HsExplicit"], ["1,3thiazol4amine","[NH]c1cscn1|HsExplicit"], ["Staurosporin","[H]N1Cc2ccccc2C1=O|HsExplicit"], ["7Hpyrrolo[2,3d]pyrimidine2,4diamine","Nc1nc(N)c2cc[nH]c2n1|HsExplicit"], ["Oxindole","O=C1Cc2ccccc2[NH]1|HsExplicit"], ["1Hpyrazolo[3,4b]pyridine","c1n[nH]c2ncccc12|HsExplicit"], ["7Hpyrrolo[2,3d]pyrimidin2amine","Nc1ncc2cc[nH]c2n1|HsExplicit"], ["2-Aminothiazole","[NH]c1nccs1|HsExplicit"], ["imidazo[1,2a]pyrazin8amine","[NH]c1nccn2ccnc12|HsExplicit"], ["Benzamidazole","c1nc2ccccc2[nH]1|HsExplicit"], ["Amitrole","Nc1nc[nH]n1|HsExplicit"], ["pyrazolo[1,5a]pyrimidin7amine","[NH]c1ccnc2ccnn12|HsExplicit"], ["3aminoindazole","[NH]c1n[nH]c2ccccc12|HsExplicit"], ["pyrimidone", "[H]N1C=NC(a)=C(a)C1=O|HsExplicit"], ["arylpyrazole_triazole", "[H][#7]-1-[#6,#7]=[#6]-[#6](-a)=[#6,#7]-1|HsExplicit"], ["maleimide","O=C1[NH]C(=O)C=C1|HsExplicit"] ] def improve_pattern(pat): pat = pat.replace("=[#16]", "=S") pat = pat.replace("=[#8]", "=O") # pat = pat.replace("-[#1]", "-[H]") pat = pat.replace("[Cl]", "Cl") pat = pat.replace("[F]", "F") pat = pat.replace("[Br]", "Br") # pat = pat.replace("[#1]", "[H]") pat = pat.replace("[#17]", "Cl") pat = pat.replace("[#9]", "F") pat = pat.replace("[#35]", "Br") pat = pat.replace("-[#16]-", "-S-") pat = pat.replace("-[#8]-", "-O-") pat = pat.replace("-[#16](=O)(=O)-", "-S(=O)(=O)-") pat = pat.replace("-[#7]=", "-N=") pat = pat.replace("=[#7]-", "=N-") pat = pat.replace("=[#6](-", "=C(-") pat = pat.replace("-[#6](=", "-C(=") pat = pat.replace("-[#6](-[H])(-[H])-", "-C(-[H])(-[H])-") return pat patterns = [[i[0], i[1], improve_pattern(i[1])] for i in patterns] class match_multiple(ProgressRunnable): def __init__(self): # self.starttime = time.time() self.useMatchCount = 0 self.calcSMILES = False self.nostructure = False self.structureColumn = vtable.findColumnWithName("SMILES") if self.structureColumn == None: self.calcSMILES = True #vortex.alert(str(self.calcSMILES)) #vortex.alert(str(vtable.findColumnWithName(vtable.MolfileColumn))) if (self.calcSMILES == True ) & (vtable.findColumnWithName(vtable.MolfileColumn) == None): vortex.alert("You need an SD file or a SMILES column") self.nostructure = True def doCalcSmiles(self): self.structureColumn.setValueFromString(vtable.getRealRowCount() - 1, None) q = Queue(processorcount * 20) #The workers t = [] #Create workers for i in range(0, processorcount): t.append(smilesworker(q, self.structureColumn)) #Start the workers for i in range(0, processorcount): t[i].start() #Load the Q for row in range(0, vtable.getRealRowCount()): q.put(row) #Something to sell the workers to stop for i in range(0, processorcount): q.put(None) for i in range(processorcount): t[i].join() def updateProgress(self, perc, message): self.setProgressValue(perc) self.setProgressMessage(message) def run(self): if not self.nostructure: self.updateProgress(0, 'Calculating SMILES') if (self.calcSMILES): self.structureColumn = vtable.findColumnWithName("SMILES", 1, vortex.STRING) self.doCalcSmiles() self.updateProgress(0, 'Indexing SMILES (for performance)') Mol2Img.doSearch(self.structureColumn, '[U].Cl.F.Br.N.O.S', 'nomdl', 0) results = ['' for i in range(0, vtable.getRealRowCount())] for i in range(0, len(patterns)): self.updateProgress(int(100 * (float(i) / float(len(patterns)))), patterns[i][0]) hits = Mol2Img.doSearch(self.structureColumn, patterns[i][2], 'nomdl', 0) mylist = hits.keySet().toArray() for j in range(0, len(mylist)): if results[mylist[j]] == '': results[mylist[j]] = patterns[i][0] else: results[mylist[j]] = results[mylist[j]] + ',' + patterns[i][0] self.resultCol = vtable.findColumnWithName('Potential Kinase', 1, vortex.STRING) self.resultCol.setValuesFromStrings(results) vtable.fireTableStructureChanged() vtable.fireTableStructureChanged() if vws is None: vortex.alert("You must have a workspace loaded...") else: matcher = match_multiple() vortex.run(matcher, "Generating matches") |
The script can be downloaded from here.
Last updated 13 March 2018