Clustering is an invaluable cheminformatics technique for subdividing a typically large compound collection into small groups of similar compounds. One of the advantages is that once clustered you can store the cluster identifiers and then refer to them later this is particularly valuable when dealing with very large datasets. This often used in the analysis of high-throughput screening results, or the analysis of virtual screening or docking studies.
In theory any molecular descriptor can be used in clustering, in practice the use of a large number of descriptors, in particular some of the more esoteric ones, can make it difficult for a medicinal chemist to understand why compounds might be in the same cluster. It is perhaps better to stick to molecular fingerprints based on structural fragments, this usually results in clusters that chemists can see are similar. For this work I’ve tried to stick with Morgan/Circular type descriptors, however since all the packages will have their own implementation they are probably not exactly the same. A number of clustering techniques are available but the most common are nearest neighbours, this method assigns a single compound to each of the desired number of clusters, ideally the most diverse selection. Next each compound remaining is assigned to the nearest cluster, the centroid of each cluster is recalculated as each new compound is assigned to the cluster.
I’ve looked at a number of options for clustering molecules from toolkits like RDKit to commercial applications such as Vortex and I’ve tried them with a variety of different sized data sets, one containing 789 molecules, one 150,000 molecules and a large set containing 4,400,000 molecules. The clustering tools often have many different options but I’ve tried to stick to the default values. All work was run on a 12-core MacPro desktop with 64GB of RAM with 1TB of internal SSCD storage. I’m sure there are other options but I’ve limited the work to those that have in built support for chemistry. I’ve also limited the investigation to applications that are run locally since I suspect most chemists won’t want to run calculations on an external server.
RDKit
RDKit is an open source toolkit for cheminformatics, it has Core data structures and algorithms in C++ with a user friendly Python wrapper. It also integrates nicely with Jupyter Notebooks. The RDKit Cookbook contains tips for using the the Butina clustering algorithm D Butina, ‘Unsupervised Database Clustering Based on Daylight’s Fingerprint and Tanimoto Similarity: A Fast and Automated Way to Cluster Small and Large Data Sets’, JCICS, 39, 747-750 (1999) and I’ve implemented it in a Jupyter Notebook.
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 |
from rdkit import Chem from rdkit.Chem import AllChem #Define clustering setup def ClusterFps(fps,cutoff=0.2): from rdkit import DataStructs from rdkit.ML.Cluster import Butina # first generate the distance matrix: dists = [] nfps = len(fps) for i in range(1,nfps): sims = DataStructs.BulkTanimotoSimilarity(fps[i],fps[:i]) dists.extend([1-x for x in sims]) # now cluster the data: cs = Butina.ClusterData(dists,nfps,cutoff,isDistData=True) return cs |
Now read in the molecules
|
1 2 3 4 5 |
#Import molecules ms = [x for x in Chem.SDMolSupplier('ApprovedDrugs.sdf',removeHs=False)] len(ms) 789 |
This dataset contains < 1000 molecules, now generate fingerprints
|
1 2 |
fps = [AllChem.GetMorganFingerprintAsBitVect(x,2,1024) for x in ms] |
Then cluster
|
1 2 |
clusters=ClusterFps(fps,cutoff=0.4) |
For a modest dataset of <1000 molecules the clustering is almost instantaneous , we can then look at a specific cluster
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 |
#show one of the clusters print(clusters[20]) (553, 297, 547) #now display structures from one of the clusters from rdkit.Chem import Draw from rdkit.Chem.Draw import IPythonConsole m1 = ms[553] m2 = ms[297] m3 = ms[547] mols=(m1,m2,m3) Draw.MolsToGridImage(mols) |

Using the larger dataset of 150,000 molecules, reading in the structures took 90 seconds and 3.8 GB RAM, adding the fingerprints took another 20 secs and took RAM usage up to 3.83GB. Clustering used only one core, and after 23 mins and consuming 80GB RAM the kernel died.
After chatting to Greg Landrum, he suggested if using a large set of molecules you wouldn’t want to ever have all the molecules in memory, so it might be better to read the molecules and generate the fingerprints in a single step like this
|
1 2 |
fps = [AllChem.GetMorganFingerprintAsBitVect(x,2,2048) for x in Chem.ForwardSDMolSupplier('ApprovedDrugs.sdf',removeHs=False) if x is not None] |
Also for the clustering use
|
1 2 |
cs = Butina.ClusterData(dists,nfps,cutoff,isDistData=True, reordering=True) |
The reordering flag is documented as follows “if this toggle is set, the number of neighbors is updated for the unassigned molecules after a new cluster is created such that always the molecule with the largest number of unassigned neighbors is selected as the next cluster center”.
Whilst this lowered the initial memory requirements, unfortunately once the clustering started the memory usage steadily increased and the kernel died.
Chemfp
Chemfp is a set of command-line tools and a Python library for fingerprint generation and high-performance similarity searches. Whilst the current version is commercial older versions are available for free and can be installed using PIP. However it seems you have to explicitly define the version to install
|
1 2 |
pip install chemfp==1.1p1 |
This installs a variety of command line tools.
The command-line tools are written in Python, with a C extension for better performance. The Python library has a large and well-documented public API which you can use from your own programs. The core functionality works with fingerprints as byte or hex strings, reads and writes fingerprint files, and performs similarity searches. Chemfp does not understand chemistry. Instead, it knows how to use RDKit, OEChem/OEGraphSim, and Open Babel to handle molecule I/O and to compute fingerprints for a molecule, and makes all of that work through a portable cross-toolkit API.
The help is incredibly well documented, for example to generate the fingerprint file we need to use rdkit2fps, typing
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 |
rdkit2fps -h usage: rdkit2fps [-h] [--fpSize INT] [--RDK] [--minPath INT] [--maxPath INT] [--nBitsPerHash INT] [--useHs 0|1] [--morgan] [--radius INT] [--useFeatures 0|1] [--useChirality 0|1] [--useBondTypes 0|1] [--torsions] [--targetSize INT] [--pairs] [--minLength INT] [--maxLength INT] [--maccs166] [--substruct] [--rdmaccs] [--id-tag NAME] [--in FORMAT] [-o FILENAME] [--errors {strict,report,ignore}] [filenames [filenames ...]] Generate FPS fingerprints from a structure file using RDKit positional arguments: filenames input structure files (default is stdin) optional arguments: -h, --help show this help message and exit --fpSize INT number of bits in the fingerprint (applies to RDK, Morgan, topological torsion, and atom pair fingerprints (default=2048) --id-tag NAME tag name containing the record id (SD files only) --in FORMAT input structure format (default guesses from filename) -o FILENAME, --output FILENAME save the fingerprints to FILENAME (default=stdout) --errors {strict,report,ignore} how should structure parse errors be handled? (default=strict) RDKit topological fingerprints: --RDK generate RDK fingerprints (default) --minPath INT minimum number of bonds to include in the subgraph (default=1) --maxPath INT maximum number of bonds to include in the subgraph (default=7) --nBitsPerHash INT number of bits to set per path (default=4) --useHs 0|1 include information about the number of hydrogens on each atom (default=1) RDKit Morgan fingerprints: --morgan generate Morgan fingerprints --radius INT radius for the Morgan algorithm (default=2) --useFeatures 0|1 use chemical-feature invariants (default=0) --useChirality 0|1 include chirality information (default=0) --useBondTypes 0|1 include bond type information (default=1) RDKit Topological Torsion fingerprints: --torsions generate Topological Torsion fingerprints --targetSize INT number of bits in the fingerprint (default=4) RDKit Atom Pair fingerprints: --pairs generate Atom Pair fingerprints --minLength INT minimum bond count for a pair (default=1) --maxLength INT maximum bond count for a pair (default=30) 166 bit MACCS substructure keys: --maccs166 generate MACCS fingerprints 881 bit substructure keys: --substruct generate ChemFP substructure fingerprints ChemFP version of the 166 bit RDKit/MACCS keys: --rdmaccs generate 166 bit RDKit/MACCS fingerprints This program guesses the input structure format based on the filename extension. If the data comes from stdin, or the extension name us unknown, then use "--in" to change the default input format. The supported format extensions are: File Type Valid FORMATs (use gz if compressed) --------- ------------------------------------ SMILES smi, ism, can, smi.gz, ism.gz, can.gz SDF sdf, mol, sd, mdl, sdf.gz, mol.gz, sd.gz, mdl.gz |
Generating the fingerprint file
|
1 2 |
rdkit2fps /Users/Chris/Desktop/Clustering/random150kmols.sdf --morgan --radius 2 > /Users/Chris/Desktop/Clustering/random150kmols.fps |
Andrew Dalke has provided a python script to help with the clustering and give some information on timings etc. it is available from here
Running on the ApprovedDrugs 789 dataset with a similarity threshold of 0.6 was effectively instantaneous
|
1 2 |
python ~/Scripts/Python/taylor_butina.py --profile --threshold 0.6 /Desktop/Clustering/rapprovedDrugs.fps -o /Desktop/Clustering/rapprovedDrugs.clusters |
The resulting cluster file can be opened in a text editor and has this format
|
1 2 3 4 5 6 7 |
Raltegravir has 51 other members => Caffeine Enprofylline Chlorzoxazone Riboflavin Ceftaroline fosamil Mycophenolate mofetil Pemirolast Paroxetine Carbetocin Triptorelin Sertindole Niclosamide Flutemetamol (18F) Theophylline Chloroxine Celecoxib Tavaborole Diiodohydroxyquinoline Nilutamide Aztreonam Tinidazole Trimetrexate Theobromine Papaverine Vandetanib Anastrozole Omeprazole Enzalutamide Fluconazole Trimethadione Bazedoxifene Gefitinib Nizatidine Aprepitant Risperidone Trypan blue Trioxsalen Almitrine Probucol Thymol Polythiazide Pazopanib Epirizole Tedizolid Phosphate Forasartan Furazolidone Nitrofurantoin Pyridoxine Dasabuvir Lapatinib Regorafenib Cilostazol has 29 other members => Quinine Lorpiprazole Tacrolimus Trimetazidine Milrinone Busulfan Micafungin Brexpiprazole Rizatriptan Calcidiol Pirlindole Bosutinib Piperonyl butoxide Metocurine Chlorothiazide Hydrochlorothiazide Florbetaben (18F) Mercaptopurine Azathioprine Finafloxacin Adenine Desloratadine Sodium lauryl sulfate Flucytosine MACITENTAN Griseofulvin Allopurinol Riluzole Cholecalciferol Dexmedetomidine has 15 other members => Fomepizole Butalbital Rilpivirine Trimethoprim Methimazole Florbetapir (18F) Cimetidine Dalfampridine Sulconazole Aprobarbital Etravirine Thiabendazole Brimonidine Tioguanine |
With some Python scripting it should be straight-forward to edit the script to output to cluster numbers and combine them with the original sdf file that was used to generate the fingerprint file.
Running the same process with a larger dataset took an unexpectedly long time and I noticed that it was only using one core. Chatting with Andrew Dalke it was apparent that Clang, the “LLVM native” C/C++/Objective-C compiler, under MacOSX, doesn’t include OpenMP support. Chemfp uses OpenMP for parallelism.
Compiling Chemfp for OpenMP
To compile Chemfp for OpenMP you’ll need to install gcc then use the CC environment variable to configure the build step to use that compiler. I’ve written up instructions for installing gcc (plus other things) hereusing Homebrew.
I had to first remove Chemfp
|
1 2 |
pip uninstall chemfp |
However, you’ll have to remove your local pip wheel file. A “wheel” file is the new(ish) Python distribution format. It can include pre-compiled code, including byte code. “pip install” internally makes a wheel file, stores it in a cache, then installs from the wheel file. The “pip uninstall” will remove the installation but notthe wheel file.
|
1 2 |
rm -rf ./Library/Caches/pip/wheels/ee/78/c6/57ed684184050a458fe0df5f4eb4ef2c50c801c6002278f191/chemfp-1.1p1-cp27-cp27m-macosx_10_12_x86_64.whl |
Then reinstall Chemfp
|
1 2 |
env CC=gcc-6 pip install chemfp==1.1p1 |
I then ran the same exercise with the 150,000 structure file, this time running at similarities of 0.8 and 0.6 to see how it might effect performance, generating the fingerprint file took very little time and the timings for the clustering are shown below.
|
1 2 |
python ~/Scripts/Python/taylor_butina.py --profile --threshold 0.8 /Users/Chris/Desktop/Clustering/random150kmols.fps -o /Users/Chris/Desktop/Clustering/random150kmols.clusters |
took just under a minute for 0.8 threshold and made use of all available cores.
|
1 2 |
python ~/Scripts/Python/taylor_butina.py --profile --threshold 0.6 /Users/Chris/Desktop/Clustering/random150kmols.fps -o /Users/Chris/Desktop/Clustering/random150kmols.clusters |
took just over a minute at 0.6, so a little slower. As before the cluster output was a text file with the same format.
I had some problems generating the fingerprint file for the 4.4 million structures, it is often a problem when a file is derived from mulitiple sources and quality control is not consistent.
I ended up tidying up the file using this script.
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 |
#!/usr/bin/env python # -*- coding: utf-8 -*- """ sdfID2smi.py ~~~~~~~~~~~~~~ Read SDF files and output SMILES for each molecule ignoring molecules that cause errors. Usage python sdfID2smi.py path/to/Compound_*.sdf.gz > smiles.txt 2> log.txt """ from __future__ import print_function from __future__ import unicode_literals from __future__ import division import gzip import sys from rdkit import Chem def readSdf(filename): print('%s\nReading file: %s\n%s' % ('='*100, filename, '-'*100), file=sys.stderr) success, fail = 0, 0 for rdmol in Chem.ForwardSDMolSupplier(gzip.open(filename)): if rdmol is None: print(' => Failed to read molecule', file=sys.stderr) fail += 1 continue try: smiles = Chem.MolToSmiles(rdmol, isomericSmiles=True) except (ValueError, RuntimeError): print(' => Failed to generate SMILES', file=sys.stderr) fail += 1 continue success += 1 print('%s\t%s' % (smiles, rdmol.GetProp('ID'.encode()))) print('%s\nSummary for: %s' % ('-'*100, filename), file=sys.stderr) print(' => %s successes, %s failures\n%s\n\n\n' % (success, fail, '='*100), file=sys.stderr) if __name__ == '__main__': if len(sys.argv) < 2: print('Usage: %s file.sdf.gz' % sys.argv[0], file=sys.stderr) sys.exit(1) for filename in sys.argv[1:]: readSdf(filename) |
This gave me a data set in SMILES format of just over 4.3 million structures. I then regenerated the sdf file and used that to generate the fingerprints without incident. (Apparently you can add “–errors ignore” to the rdkit2fps command, I’ve not tested it).
Generating the fingerprint file for the 4.3 million structures took 35 mins, generating the clusters using a 0.8 similarity took 10 hours. The python script also outputs a useful summary.
|
1 2 3 4 5 6 |
#fingerprints: 4333045 #bits/fp: 2048 threshold: 0.8 #matches: 10009412 Load time: 29.5 sec memory: 1.00 GB Similarity time: 35825.8 sec memory: 1.00 GB Clustering time: 14.3 sec memory: 733 MB Total time: 35884.5 sec |
DataWarrior
DataWarrior is an open-source data analysis package I’ve reviewed previously. The cluster algorithm implemented in DataWarrior is simple, reproducible, but computationally demanding. By default Datawarrior uses Fragfp fingerprints
FragFp is a substructure fragment dictionary based binary fingerprint similar to the MDL keys. It relies on a dictionary of 512 predefined structure fragments.
To get a better comparison with the other clustering algorithms I generated the SphereFp fingerprint.
SphereFp descriptor encodes circular spheres of atoms and bonds into a hashed binary fingerprint of 512 bits. From every atom in the molecule, DataWarrior constructs fragments of increasing size by including n layers of atom neighbours (n=1 to 5). These circular fragments are canonicalized considering aromaticity, but neglecting stereo configurations. From the canonical representation a hash code is generated, which is used to set the respective bit of the fingerprint.
One the dataset of 789 molecules the clustering was instantaneous, and it was very easy to browse through the results.

Reading in the 150,000 structure file took 20 seconds and consumed 550 MB of RAM, adding the descriptors took the RAM usage up to 1.14GB. Unfortunately when I tried to do the clustering I got this message.

MOE
MOE is a molecular modelling package from Chemical Computing Group. Whilst MOE does not include Morgan/circular type fingerprints there is a SVL script based on the work by Rogers et al DOI that can be used to generate them. One the small dataset of 789 structures the clustering was complete within 15 seconds. One the larger set of 150,000 molecules generation of the fingerprints took 2 mins and the subsequent clustering took (>40 hours and counting)
I did not attempt to cluster the 4.4 million structure file using MOE.
Vortex
Vortex is a high performance, interactive data visualization and analysis application, with native cheminformatics and bioinformatics intelligence built in. Reading in the 789 structure file and then clustering was instantaneous. Vortex provides k-means clustering as standard, and uses either atom-based similarity (called DotFPCA) hex-packed to 1024 bits. This is the Dotmatics implementation of Morgan/circular type fingerprints.

Working with the 150,000 structure file, again using the default options, took less than a minute to generate the fingerprints and complete the clustering.
Using the 4.4M structure file took a little longer, reading in the file took less than 3 mins and consumed 13GB of RAM. Generating the fingerprints took 18 mins. used 4 cores and took the RAM usage up to 16GB, the clustering then took a further 40 mins using available cores with the RAM usage rising to 17.5 GB. Two columns are added: a CLUSTER number, and a DISTANCE; the centroid has a distance of 0 and all other compounds vary in distance up to 1.
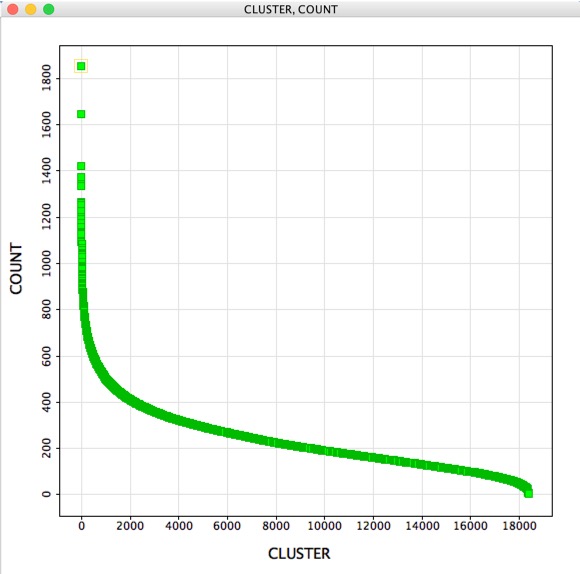
With over 4 million structures it is not really practical to simply scroll through the table, but here are a couple of tools that might help with further analysis. A question that might then arise is “How many molecules belong to each cluster?”, the GenericClusterAnalysis script creates a new workspace containing two columns. The first containing the cluster number the second the count of occurrences for each cluster, for this 4.4 million structures it took a couple of seconds to run.


Alternatively you can use a variation on the duplicate check script to select the first example from each cluster, you can download it here http://macinchem.org/reviews/vortexscripts/ChoseOneFromClusters.vpy.zip. An alternative is to select the centroid of each cluster by modifying the script to perform an additional selection based on the DISTANCE value, the script can be downloaded here http://macinchem.org/reviews/vortexscripts/ChoseCentreFromClusters.vpy.zip.

Dan Ormsby at Dotmatics provided me with a couple of script snippets that expose the fingerprint generation code, it turns out that under the hood there are a wide variety of fingerprint options the default options are all pre-set for the typical medicinal chemist user. I used the 150,000 molecule set to explore the impact of varying a couple of the options. Changing the atom path length in the fingerprint from 2 to 10 had minimal impact on the time taken, all less than 1 minute. To improve efficiency when working with these fingerprints they are usually delivered into Vortex in a hex packed format, the default is 1024 bits. I also did the clustering using the unpacked fingerprints since this would represent the slowest method. Using the unpacked fingerprints the run took two minutes to complete.
KNIME
There is also a RSC CICAG workshop describing the use of KNIME for clustering.
Summary
For small data sets of a few thousand structures there are multiple options for clustering, from Open Source toolkits to sophisticated desktop applications. However as the dataset increases the computational demands increase, and support for the use of all available cores becomes essential. For very large datasets I’ve only found two options Chemfp and Vortex. I’ve only used the free version of Chemfp (the commercial version may have better performance) and whilst it copes with several million structures it is very considerably slower than Vortex. Vortex also has the advantage of having the nice graphical user interface and scripting tools to allow further exploration of the dataset.
Last Updated 22 July 2017
2 thoughts on “Options for Clustering large datasets of Molecules”
Comments are closed.