One of the most common tasks for those involved in cheminformatics is handling files containing molecular information, these files can be in a variety of file types and usually the task involved is relatively minor. cApp is Java application that provides a simple interface to a variety of everyday activities.
cApp requires JRE7 and uses the Chemistry Development Kit (CDK), an open-source Java library for chem- and bioinformatics, and associated software, JChemPaint as chemical editor, and routines developed within the Program Collection for Structural Biology and Biophysical Chemistry by the Hofmann group. Full details of cApp are described in a J Cheminformatics paper DOI.
Starting cApp
You can start the GUI application by double clicking on the downloaded jar file, or from the Terminal using
|
1 |
java -jar {path to capp.jar file} [switches] |
The command line options allows you complete specific asks without invoking the GUI.
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 |
java -jar /Users/swain/Desktop/capp_v1.3_java1.7.jar -h cApp v1.3 - Help cApp - A Java tool for compound appraisal USAGE java -jar {capp.jar file} [switches] TASKS appraise Compound appraisal with calculation of descriptors and PAINs similarity. cluster Group compounds into clusters based on Tanimoto similarity. smsd Similarity search of compound(s) against libraries by maximal common subgraph. split Split a library into subsets (only for SDF libraries) SWITCHES -appraise Runs the appraise task (default). -ascii Write results in ASCII format. -autoselect Auto-select largest entity when reading SDF or SMILES. -cluster {N} Group compounds into {N} clusters. -debug Debug option. -drug Evaluate for drug likeness (Lipinski's Rule of 5). -duplicates inchi Check for duplicate entries based on InChI Keys. -duplicates tanimoto Check for duplicate entries based on Tanimoto similarity. -fragment Evaluate for fragment likeness (Rule of 3). -gui Start the program with the graphical user interface. -h Print this help. -html Generate results in HTML format. -i {input file} Input file with compounds to process. -inchi Input file contains InChI code. -lead Evaluate for lead likeness. -load Load a previously saved cApp project. -maxsets {N} Maximum number of compound sets in the project (default: 10). -pdf Write results into PDF files. -pdf landscape PDF paper orientation is Landscape (default). -pdf portrait PDF paper orientation is Portrait. -pdf bondlength Structure images are drawn with same bond length (default). -pdf fixed Structure images have the same fixed size. -png Write PNG images of compounds. -pubchem Search for entry in PubChem when conducting the appraise task. -sdf Input file is an SDF file. -smi Input file contains SMILES code. -smsd {library file} Similarity search of {input file} against {library file} in SDF format. -split {N} Split an SDF library into subsets of {N} entries each. -svg Write SVG images of compounds. -3d Attempt to generate 3D coordinates. |
One command that might be particularly useful is -split, the developers suggest that users avoid working with compound sets of greater than 1000 molecules, large library files in SDF format can be split using the split task. Also similarity searches that use large libraries can be carried out when starting the task from the terminal without the GUI using the -smsd switch.
Using the GUI
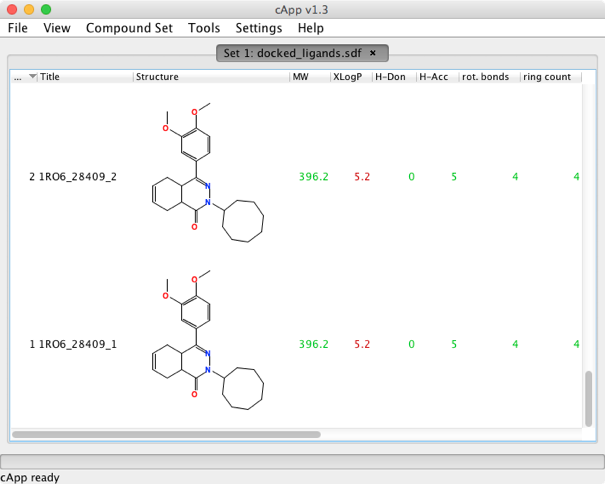
Double clicking on the jar file open a blank window, for Mac users this may look a little strange since the menus are attached to the window not the top menu bar. To import molecules simply select “Add compounds as new set” from the file menu (SMILES, INChi or SDF format). The import is reasonably quick for small file sizes, I also tested opening an sdf containing 993 molecules and 20 fields, this took over 30 mins. A number of physicochemical properties are automatically calculated on import (MW, XLogP, HBD, HBA etc.). These properties are calculated locally using the CDK toolkit. The settings tab can be used to set the default to auto-select the largest entity when reading SDF or SMILES, this strips salts and counter-ions on import.
Once imported you can choose which columns to hide/view, the columns can also be sorted but it seems you cannot change the order of columns, all actions are pretty responsive so interactive analysis and viewing a perfectly fine on modest datasets. It is worth noting that only the columns set as visible will be written into output files.
Viewers/Editors
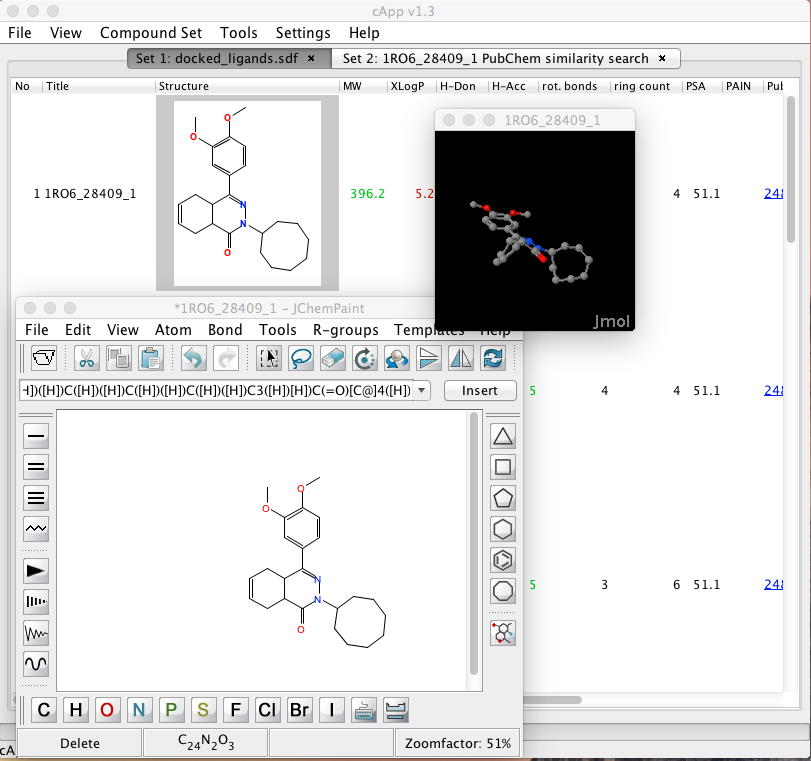
Right-clicking on a structure brings up a contextual menu that allows you transfer the structure to a molecule editor (JChemPaint), or generate a 3D structure and view it in Jmol, the viewer only allows display and right-clicking on the Jmol window does not give access to the usual Jmol functionality, and you can only have a single 3D structure displayed at a time although the authors are gathering feedback about what features users might like to access from the Jmol window. Note that when closing the JChemPaint session with Accept, the selected compound entry will be updated with the modified structure; this will result in a 2D structure.
You can also view the meta data associated with a structure and calculate a variety of properties.
Calculations
The tools menu gives access to a variety of calculations and services,
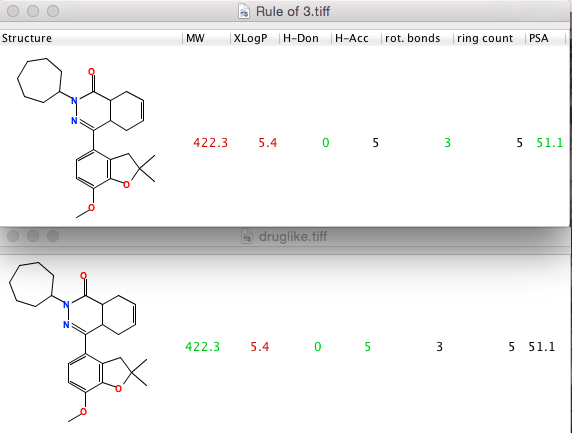
Likeness analysis colour codes the calculated properties based on the likeness profile chosen Drug (Rule of 5) , Lead (Leadlike) or Fragment (Rule of 3), the image below compares the fragment v drug likeness. If the criteria for the selected likeness are met, the values are highlighted in green; if they are not met, the values are shown in red. If they are black, the property is not part of the chosen rule set.
Searching
cApp provides some chemical searching capableilities, first select a compound by a left-click on any cell in the row of the desired compound. Then right-click to obtain the pop-up menu, the PubChem searches send the query as a SMILES string over the internet so don’t use this for confidential material.
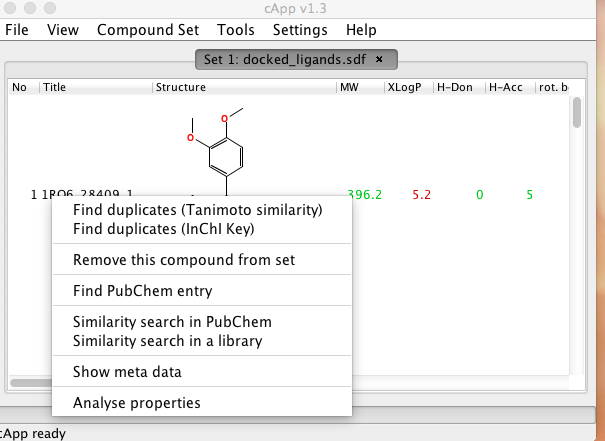
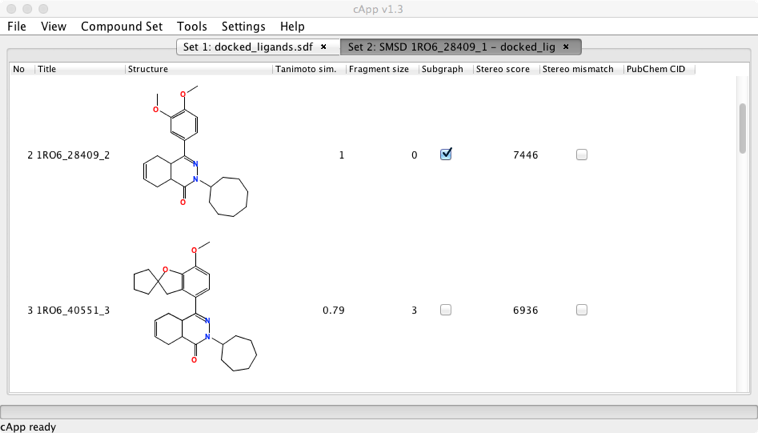
The “Similarity search in a library” option allows you to search a user provided file (in SDF format), the results are displayed in a new tab together with the Tanimoto similarity score. It is worth noting that cApp will process the entire provided library and so for large files this can become very slow. Similarity searches that use large libraries can be also be carried out by starting the task from the terminal without the GUI using the -smsd switch.
Writing Files
You can save results as a cApp project, this has the .cpp extension which unfortunately also happens to be the extension used by the C++ programming language, this means by default if you save on the saved project file it will probably open in Xcode. If you right-click on the file you can use the “Open with” option. In addition to the native .cpp format, results can also be written as SDF, SMILES or InChI/InChI Key format. Results can also be stored as images or PDF/HTML, of course much of the chemical information will be lost in these formats.
Summary
cApp serves a very important niche, where scientists need to quickly manipulate small lists of molecules. It does this task very nicely, however if you are going to be routinely handling/analysing larger datasets it is probably worth investing in an alternative application.
Last Updated 3 July 2015